
WEGOVY 1,7 mg, FlexTouch solution injectable en stylo prérempli, boîte de 1 stylo prérempli ( 4 aiguilles) de 3 ml
Dernière révision : 14/10/2024
Taux de TVA : 10%
Laboratoire exploitant : NOVO NORDISK
Source :

Adultes
Wegovy est indiqué en complément d'un régime hypocalorique et d'une augmentation de l'activité physique dans le contrôle du poids, notamment pour la perte de poids et le maintien du poids, chez des adultes avec un indice de masse corporelle (IMC) initial de :
- ≥ 30 kg/m2 (obésité), ou
- ≥ 27 kg/m2 et < 30 kg/m2 (surpoids) en présence d'au moins un facteur de comorbidité lié au poids tel qu'une dysglycémie (prédiabète ou diabète de type 2), une hypertension artérielle, une dyslipidémie, un syndrome d'apnée obstructive du sommeil ou une maladie cardiovasculaire.
Pour les résultats des essais concernant la réduction du risque cardiovasculaire, l’insuffisance cardiaque liée à l'obésité et les populations étudiées, voir rubrique Propriétés pharmacodynamiques.
Adolescents (≥ 12 ans)
Wegovy est indiqué en complément d'un régime hypocalorique et d'une augmentation de l'activité physique dans le contrôle du poids chez des adolescents âgés de 12 ans et plus avec :
- une obésité* et
- un poids corporel supérieur à 60 kg.
Le traitement par Wegovy doit être arrêté et réévalué si les patients adolescents n'ont pas réduit leur IMC d'au moins 5 % après 12 semaines à la dose de 2,4 mg ou à la dose maximale tolérée.
*Obésité (IMC ≥ 95e percentile) défini selon les courbes de croissance de l'IMC en fonction du sexe et de l'âge (CDC.gov) (voir Tableau 1).
Tableau 1 Seuils d'IMC pour l'obésité (≥ 95e percentile) par sexe et par âge pour les patients pédiatriques âgés de 12 ans et plus (critères CDC)
| Age (années) | IMC (kg/m2) au 95e percentile | |
| Hommes | Femmes | |
| 12 | 24,2 | 25,2 |
| 12,5 | 24,7 | 25,7 |
| 13 | 25,1 | 26,3 |
| 13,5 | 25,6 | 26,8 |
| 14 | 26,0 | 27,2 |
| 14,5 | 26,4 | 27,7 |
| 15 | 26,8 | 28,1 |
| 15,5 | 27,2 | 28,5 |
| 16 | 27,5 | 28,9 |
| 16,5 | 27,9 | 29,3 |
| 17 | 28,2 | 29,6 |
| 17,5 | 28,6 | 30,0 |
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Aspiration pulmonaire en association avec une anesthésie générale ou une sédation profonde
Des cas d'aspiration pulmonaire du contenu gastrique ont été signalés chez des patients recevant des agonistes des récepteurs du GLP-1 subissant une anesthésie générale ou une sédation profonde. Par conséquent, le risque accru de contenu gastrique résiduel en raison du retard de vidange gastrique (voir rubrique Effets indésirables) doit être pris en considération avant de réaliser des procédures impliquant une anesthésie générale ou une sédation profonde.
Déshydratation
L'utilisation d'agonistes des récepteurs du GLP-1 peut être associée à des effets indésirables gastro-intestinaux pouvant entraîner une déshydratation qui, dans de rares cas peuvent conduire à une détérioration de la fonction rénale. Les patients doivent être avertis du risque potentiel de déshydratation lié aux effets indésirables gastro-intestinaux et doivent prendre des précautions pour éviter une perte hydrique.
Pancréatite aiguë
Des cas de pancréatite aiguë ont été observés lors de l'utilisation d'agonistes des récepteurs du GLP-1 (voir rubrique Effets indésirables).
Les patients doivent être informés des symptômes caractéristiques de la
pancréatite aiguë. En cas de suspicion de pancréatite, le sémaglutide
devra être arrêté ; si la pancréatite est confirmée, le sémaglutide ne
devra pas être réadministré. Il convient d'être prudent chez les
patients ayant des antécédents de pancréatite.
En l'absence d'autres signes et symptômes de pancréatite aiguë, des
élévations des enzymes pancréatiques seules ne prédisent pas une
pancréatite aiguë.
Patients atteints de diabète de type 2
Le sémaglutide ne doit pas être utilisé comme substitut de l'insuline chez les patients atteints de diabète de type 2.
Le sémaglutide ne doit pas être utilisé en association avec d'autres agonistes des récepteurs du GLP-1. L'association n'a pas été étudiée et un risque accru d'effets indésirables liés à un surdosage est considéré comme probable.
Hypoglycémie chez les patients atteints de diabète de type 2
L'insuline
et les sulfamides hypoglycémiants sont connus pour provoquer une
hypoglycémie. Les patients traités par le sémaglutide en association à
un sulfamide hypoglycémiant ou à une insuline peuvent présenter une
augmentation du risque d'hypoglycémie. Le risque d'hypoglycémie peut
être diminué en réduisant la dose du sulfamide hypoglycémiant ou de
l'insuline lors de l'initiation du traitement par un agoniste des
récepteurs du GLP-1.
Rétinopathie diabétique chez les patients atteints de diabète de type 2
Un risque accru de complications liées à la rétinopathie diabétique a été observé chez les patients atteints de rétinopathie diabétique et traités par sémaglutide (voir rubrique Effets indésirables). Une amélioration rapide du contrôle glycémique a été associée à une aggravation temporaire de la rétinopathie diabétique, cependant d'autres mécanismes ne peuvent pas être exclus. Les patients atteints de rétinopathie diabétique et traités par sémaglutide doivent faire l'objet d'une surveillance attentive et doivent être traités selon les recommandations cliniques. Il n'y a pas d'expérience avec Wegovy chez les patients atteints de diabète de type 2 et présentant une rétinopathie diabétique non contrôlée ou potentiellement instable. Chez ces patients, le traitement par Wegovy n'est pas recommandé.
Populations non étudiées
La sécurité et l'efficacité de Wegovy n'ont pas été étudiées chez les patients :
- traités par d'autres produits de gestion du poids,
- atteints de diabète de type 1,
- présentant une insuffisance rénale sévère (voir rubrique Posologie et mode d'administration),
- présentant une insuffisance hépatique sévère (voir rubrique Posologie et mode d'administration),
- présentant une insuffisance cardiaque congestive de classe IV selon la New York Heart Association (NYHA).
L'utilisation chez ces patients n'est pas recommandée.
L'expérience de Wegovy est limitée chez les patients :
- âgés de 85 ans ou plus (voir rubrique Posologie et mode d'administration),
- présentant une insuffisance hépatique légère ou modérée (voir rubrique Posologie et mode d'administration),
- présentant une maladie inflammatoire de l'intestin,
- présentant une gastroparésie diabétique.
Le médicament doit être utilisé avec prudence chez ces patients.
Teneur en sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Dans quatre essais de phase 3a, 2 650 patients adultes ont été exposés à Wegovy. La durée des études était de 68 semaines. Les effets indésirables les plus fréquemment signalés étaient des troubles gastro- intestinaux comprenant nausées, diarrhées, constipation et vomissements.
Liste tabulée des effets indésirables
Le tableau 3 répertorie les effets indésirables identifiés dans les études cliniques chez les adultes et les rapports post-commercialisation. Les fréquences sont basées sur un ensemble d'études de phase 3a.
Les effets indésirables associés à Wegovy sont indiqués ci-dessous par classe de systèmes d'organes et par fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 3
Fréquence des effets indésirables du sémaglutide
|
MedDRA Classe de systèmes d'organes |
Très fréquent | Fréquent | Peu fréquent | Rare |
Fréquence indéterminée |
|
Affections
du système immunitaire |
Réaction
anaphylactique |
||||
|
Troubles
du métabolisme et de la nutrition |
Hypoglycémie
chez les patients atteints de diabète de type 2a |
||||
|
Affections
du système nerveux |
Maux de têteb |
Vertigesb
Dysgueusieb,c Dysesthésiea |
|||
|
Affections
oculaires |
Rétinopathie
diabétique chez les patients atteints de diabète de type 2a |
||||
|
Affections
cardiaques |
Hypotension
Hypotension orthostatique Augmentation du rythme cardiaquea,c |
||||
|
Affections
gastro- intestinales |
Vomissementsa,b
Diarrhéesa,b Constipationa,b Nauséesa,b Douleurs abdominalesb,c |
Gastriteb,c
Reflux gastro- œsophagienb Dyspepsieb Éructationb Flatulenceb Distension abdominaleb |
Pancréatite
aiguëa Retard de la vidange gastrique |
|
Obstruction
intestinale |
|
Affections
hépatobiliaires |
|
Lithiase biliairea |
|
|
|
|
Affections
de la peau et du tissu sous-cutané |
|
Perte
des cheveuxa |
|
Angioœdème |
|
|
Troubles
généraux et anomalies au site d'administration |
Fatigueb,c |
Réactions
au site d'injectionc |
|
|
|
| Investigations |
|
|
Élévation
de l'amylasec Élévation de la lipasec |
|
|
a) Voir description de
certains effets indésirables ci-dessous
Description de certaines réactions indésirables
Sauf indication contraire, les informations ci-dessous relatives à certaines réactions indésirables concernent les essais de phase 3a.
Réactions
indésirables gastro-intestinales
Sur la période
d'étude de 68 semaines, des nausées sont survenues chez 43,9 % des patients
traités par sémaglutide (16,1 % pour le placebo), des
diarrhées chez 29,7 % (15,9 % pour le placebo) et des vomissements chez 24,5 %
(6,3 % pour le placebo). La plupart des événements étaient d'intensité légère à
modérée et de courte durée. La constipation est survenue chez 24,2 % des
patients traités par sémaglutide (11,1 % pour le
placebo) et était d'intensité légère à modérée et de durée plus longue.
Chez les
patients traités par sémaglutide, la durée médiane
des nausées était de 8 jours, des vomissements de 2 jours, de la diarrhée de 3
jours et de la constipation de 47 jours.
Les patients insuffisants rénaux modérés (DFGe ≥ 30 à < 60 ml/min/1,73 m2) peuvent présenter davantage d’effets gastro-intestinaux lorsqu’ils sont traités par sémaglutide.
Les événements gastro-intestinaux ont conduit à l'arrêt définitif du traitement chez 4,3 % des patients.
Pancréatite
aiguë
La fréquence
rapportée des pancréatites aiguës confirmées par adjudication dans les études
cliniques de phase 3a était respectivement de 0,2 % pour le sémaglutide
et de < 0,1 % pour le placebo. Dans l'essai de morbi-mortalité
cardiovasculaire SELECT, la fréquence des pancréatites aiguës confirmées par
adjudication était de 0,2 % pour le sémaglutide et de
0,3 % pour le placebo.
Maladie
aiguë de la vésicule biliaire/cholélithiase
Une cholélithiase a été rapportée chez 1,6 % des patients et a
conduit à une cholécystite chez 0,6 % des patients traités par sémaglutide. Une cholélithiase et
une cholécystite ont été rapportées chez 1,1 % et 0,3 %, respectivement, des
patients traités par placebo.
Perte
des cheveux
Une perte de
cheveux a été rapportée chez 2,5 % des patients traités par sémaglutide
et chez 1,0 % des patients traités par placebo. Les événements étaient
principalement d'intensité légère et la plupart des patients se sont rétablis
lors de la poursuite du traitement. La perte de cheveux était plus souvent
rapportée chez les patients présentant une plus grande perte de poids (≥
20 %).
Augmentation de la fréquence cardiaque
Dans
les essais de phase 3a, une augmentation moyenne de 3 battements par minute (bpm) par rapport à une valeur moyenne à l'inclusion de 72 bpm a été observée chez les patients traités par sémaglutide. Les proportions de patients avec une
augmentation des pulsations par rapport à l'inclusion ≥ 10 bpm à tout moment au cours de la période de traitement
étaient de 67,0 % dans le bras sémaglutide vs 50,1 %
dans le bras placebo.
Immunogénicité
Compte tenu
des propriétés potentiellement immunogènes des médicaments contenant des
protéines ou des peptides, les patients traités par sémaglutide
peuvent développer des anticorps. La proportion de patients testés positifs aux
anticorps anti-sémaglutide à tout moment après
l'inclusion était faible (2,9 %) et aucun patient ne présentait d'anticorps
neutralisants anti-sémaglutide ni d'anticorps anti- sémaglutide avec un effet neutralisant du GLP-1 endogène à
la fin de l'essai. Pendant le traitement, des concentrations élevées de sémaglutide pourraient avoir diminué la sensibilité des
dosages, donc le risque de faux négatifs ne peut être exclu. Toutefois, chez
les patients testés positifs pour les anticorps pendant et après le traitement,
la présence des anticorps était transitoire et sans impact apparent sur
l'efficacité et la sécurité.
Hypoglycémie
chez les patients atteints de diabète de type 2
Dans l'étude
STEP 2, une hypoglycémie cliniquement significative a été observée chez 6,2 %
(0,1 événement/patient-année) des patients traités par sémaglutide,
par rapport à 2,5 % (0,03
événement/patient-année) des patients traités par placebo. Une hypoglycémie
sous sémaglutide a été observée tant avec que sans
usage concomitant de sulfamide hypoglycémiant. Un épisode (0,2 % des patients,
0,002 événement/patient-année) a été signalé comme étant sévère chez un patient
non traité concomitamment par un sulfamide hypoglycémiant. Le risque
d'hypoglycémie était accru lorsque le sémaglutide
était utilisé avec un sulfamide hypoglycémiant.
Dans l’essai STEP-HFpEF-DM, une
hypoglycémie cliniquement significative a été observée chez 4,2 % des
patients des groupes sémaglutide et placebo en cas d’association à un
sulfamide hypoglycémiant et/ou à l’insuline (0,065
événement/patient-année avec le sémaglutide et 0,098
événement/patient-année avec le placebo).
Rétinopathie
diabétique chez les patients atteints de diabète de type 2
Une étude
clinique de 2 ans a étudié le sémaglutide de 0,5 mg
et de 1 mg par rapport au placebo chez 3 297 patients atteints de diabète de
type 2, présentant un risque cardiovasculaire élevé, un diabète de longue durée
et une glycémie insuffisamment contrôlée. Dans cette étude, des événements
établis de complication de rétinopathie diabétique sont survenus chez un plus
grand nombre de patients traités par sémaglutide (3,0
%) que de patients sous placebo (1,8 %). Cette observation vaut pour les
patients traités par insuline et présentant une rétinopathie diabétique connue.
La différence entre les traitements est apparue de manière précoce et a
persisté pendant toute l'étude. Dans l’étude STEP 2, des troubles de la rétine ont été
rapportés par 6,9 % des patients traités par Wegovy,
6,2 % des patients traités par sémaglutide 1 mg et
4,2 % des patients traités par placebo. La majorité des événements ont été
rapportés comme étant une rétinopathie diabétique (4,0 %, 2,7 % et 2,7 %,
respectivement) et une rétinopathie non proliférante
(0,7 %, 0 % et 0 %, respectivement).
Dysesthésie
Des événements liés à un tableau clinique d’altération de la sensation
cutanée, tels que paresthésie, peau douloureuse, peau sensible,
dysesthésie et sensation de brûlure, ont été rapportés chez 2,1 % des
patients traités par sémaglutide 2,4 mg et 1,2 % des patients traités
par placebo. Ces événements étaient d’une gravité légère à modérée et
la plupart des patients se sont rétablis tout en continuant le
traitement.
Population pédiatrique
Dans un essai clinique mené chez des adolescents âgés de 12 ans à moins de 18 ans souffrant d'obésité ou de surpoids et présentant au moins une comorbidité liée au poids, 133 patients ont été exposés à Wegovy. La durée de l'essai était de 68 semaines.
Globalement, la fréquence, la nature et la sévérité des effets indésirables chez les adolescents ont été comparables à celles observées dans la population adulte. Une cholélithiase a été rapportée chez 3,8 % des patients traités par Wegovy et 0 % des patients traités par placebo.
Aucun effet sur la croissance ou le développement pubertaire n'a été observé après 68 semaines de traitement.
Autres populations particulières
Au cours des essais SELECT et SUSTAIN 6, chez des adultes atteints de
maladie cardiovasculaire établie, le profil d’effets indésirables était
similaire à celui observé dans les essais de phase 3a de gestion du
poids.
Dans les essais HFpEF menés chez des adultes présentant une insuffisance cardiaque à fraction d’éjection préservée (HFpEF) liée à l’obésité, le profil d’effets indésirables était similaire à celui observé dans les essais de phase 3a de gestion du poids.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) et le réseau des Centres Régionaux de Pharmacovigilance, site internet : https://signalement.social-sante.gouv.fr/
DEMANDER
UNE AIDE MEDICALE EN URGENCE ET INFORMER IMMEDIATEMENT LE MEDECIN en cas de réaction allergique
sévère : symptômes tels que des difficultés respiratoires, un gonflement, la
tête qui tourne, des battements de coeur rapides, de la transpiration
et une perte de connaissance ou un gonflement rapide sous la peau dans
les zones telles que le visage, la gorge, les bras et les jambes.
INFORMER le médecin en cas de problèmes au niveau des yeux, tels que des
modifications de la vue.
CONSULTER IMMEDIATEMENT le médecin en cas de douleurs intenses et persistantes
au niveau de l'estomac qui peuvent se déplacer dans le dos.
FEMME EN AGE DE PROCREER :
- L'utilisation d'une CONTRACEPTION est recommandée .
- Le traitement par
sémaglutide DOIT ÊTRE ARRETÉ au moins 2 mois avant un projet de
grossesse.
PRUDENCE en cas de conduite de véhicule ou
d'utilisation de machines (vertiges).
INFORMER le médecin en cas d'intervention chirurgicale nécessitant une
anesthésie (endormissement).
Femmes en âge de procréer
L'utilisation d'une contraception pendant le traitement par le sémaglutide est recommandée chez les femmes en âge de procréer (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Grossesse
Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Il existe des données limitées sur l'utilisation du sémaglutide chez la femme enceinte. Le sémaglutide ne doit donc pas être utilisé pendant la grossesse. En cas de projet de grossesse ou en cas de grossesse, le traitement par le sémaglutide doit être interrompu. Le sémaglutide doit être arrêté au moins 2 mois avant un projet de grossesse en raison de sa longue demi-vie (voir rubrique Propriétés pharmacocinétiques).
Allaitement
Le sémaglutide a été excrété dans le lait de rates allaitantes. Un risque pour l'enfant allaité ne pouvant pas être exclu, le sémaglutide ne doit pas être utilisé pendant l'allaitement.
Fertilité
L'effet du sémaglutide sur la fertilité humaine est inconnu. Le sémaglutide n'a pas affecté la fertilité des rats mâles. Chez le rat femelle, une prolongation du cycle œstral et une faible baisse du nombre d'ovulations ont été observées à des doses associées à une réduction du poids maternel.
Le sémaglutide retarde la vidange gastrique et pourrait potentiellement influencer l'absorption des médicaments administrés de façon concomitante par voie orale. Aucun effet cliniquement pertinent sur la vitesse de la vidange gastrique n'a été observé avec le sémaglutide de 2,4 mg, probablement en raison d'un effet de tolérance. Le sémaglutide doit être utilisé avec prudence chez les patients recevant des médicaments par voie orale nécessitant une absorption gastro-intestinale rapide.
Paracétamol
Le sémaglutide retarde la vidange gastrique telle qu'évaluée par la pharmacocinétique du paracétamol pendant un repas test standard. L'ASC0-60min et la Cmax du paracétamol ont baissé de 27 % et 23 % respectivement, après une utilisation concomitante de sémaglutide de 1 mg. L'exposition totale au paracétamol (ASC0-5h) n'a pas été affectée. Aucun effet cliniquement pertinent sur le paracétamol n'a été observé avec le sémaglutide. Aucun ajustement de la dose de paracétamol n'est nécessaire en cas d'association avec sémaglutide.
Contraceptifs hormonaux
Le sémaglutide ne devrait pas réduire l'effet des contraceptifs oraux. Le sémaglutide ne modifie pas de manière cliniquement significative l'exposition totale à l'éthinylestradiol et au lévonorgestrel en cas d'administration concomitante d'un contraceptif oral combiné (0,03 mg d'éthinylestradiol/0,15 mg de lévonorgestrel) avec le sémaglutide. L'exposition à l'éthinylestradiol n'a pas été affectée ; une augmentation de 20 % de l'exposition au lévonorgestrel à l'état d'équilibre a été observée. La Cmax n'a été affectée pour aucun des composés.
Atorvastatine
Le sémaglutide n'a pas modifié l'exposition totale à l'atorvastatine après administration d'une dose unique de 40 mg d'atorvastatine. La Cmax de l'atorvastatine a diminué de 38 %. Cette baisse n'a pas été considérée comme cliniquement significative.
Digoxine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la digoxine après administration d'une dose unique de 0,5 mg de digoxine.
Metformine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la metformine après administration de 500 mg deux fois par jour pendant 3,5 jours.
Warfarine et autres dérivés de la coumarine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax
de la R- et S-warfarine après une dose unique de warfarine (25 mg), et
l'effet pharmacodynamique de la warfarine tel que mesuré par le rapport
normalisé international (INR) n'a pas été affecté de manière
cliniquement significative.
Toutefois, des cas de diminution de l'INR ont été rapportés lors de
l'utilisation concomitante d'acénocoumarol et de sémaglutide. Lors de
l'initiation du traitement par sémaglutide chez des patients sous
warfarine ou autres dérivés de la coumarine, une surveillance fréquente
de l'INR est recommandée.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez les adultes.
Posologie
Adultes
La
dose d'entretien de sémaglutide de 2,4 mg une fois par semaine est
atteinte en commençant par une dose de 0,25 mg. Pour réduire la
fréquence de symptômes gastro-intestinaux, la dose doit être augmentée
sur une période de 16 semaines pour atteindre la dose d'entretien de
2,4 mg une fois par semaine (voir Tableau 2). En cas de symptômes
gastro-intestinaux significatifs, envisager de retarder l'augmentation
de la dose ou de réduire à la dose précédente jusqu'à l'amélioration
des symptômes.
Des doses hebdomadaires supérieures à 2,4 mg ne sont pas recommandées.
Tableau 2 Schéma d'augmentation de la dose
| Augmentation de la dose | Dose hebdomadaire |
| Semaines 1 à 4 | 0,25 mg |
| Semaines 5 à 8 | 0,5 mg |
| Semaines 9 à 12 | 1 mg |
| Semaines 13 à 16 | 1,7 mg |
| Dose d'entretien | 2,4 mg |
Adolescents
Pour les adolescents âgés de 12 ans et plus, le même schéma d'augmentation de la dose que celui utilisé pour les adultes doit être appliqué (voir Tableau 2). La dose doit être augmentée jusqu'à 2,4 mg (dose d'entretien) ou jusqu'à la dose maximale tolérée. Des doses hebdomadaires supérieures à 2,4 mg ne sont pas recommandées.
Patients atteints de diabète de type 2
Lors de l'initiation du traitement par sémaglutide chez des patients
atteints de diabète de type 2, une réduction de la dose d'insuline ou
des sécrétagogues de l'insuline (tels que les sulfamides
hypoglycémiants) administrés de façon concomitante doit être envisagée
afin de réduire le risque d'hypoglycémie, voir rubrique Mises en garde spéciales et précautions d'emploi.
Oubli de dose
Si une dose est oubliée, elle doit être administrée dès que possible et
dans les 5 jours suivant l'oubli. Si plus de 5 jours se sont écoulés,
la dose oubliée ne doit pas être prise, et la dose suivante doit être
administrée le jour normalement prévu. Dans chacun des cas, les
patients peuvent ensuite reprendre leur schéma posologique hebdomadaire
habituel. Si plusieurs doses sont oubliées, il convient d'envisager une
réduction de la dose de départ pour une réinstauration du traitement.
Populations particulières
Sujets âgés (≥ 65 ans)
Aucun ajustement de la dose n'est nécessaire en fonction de l'âge.
L'expérience clinique chez les patients âgés de ≥ 85 ans est limitée.
Patients insuffisants rénaux
Aucun ajustement de la dose n'est nécessaire chez les patients
présentant une insuffisance rénale légère ou modérée. L'expérience
relative à l'utilisation du sémaglutide chez des patients présentant
une insuffisance rénale sévère est limitée. Le sémaglutide n'est pas
recommandé chez les patients présentant une insuffisance rénale sévère
(DFGe < 30 ml/min/1,73 m2) y compris les patients présentant une insuffisance rénale terminale (voir rubriques Mises en garde spéciales et précautions d'emploi, Effets indésirables et Propriétés pharmacocinétiques).
Patients insuffisants hépatiques
Aucun ajustement de la dose n'est requis chez les patients présentant
une insuffisance hépatique légère ou modérée. L'expérience relative à
l'utilisation du sémaglutide chez des patients présentant une
insuffisance hépatique sévère est limitée. Le sémaglutide n'est pas
recommandé chez les patients présentant une insuffisance hépatique
sévère et doit être utilisé avec prudence chez les patients présentant
une insuffisance hépatique légère ou modérée (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
Aucun ajustement de la dose n'est nécessaire chez les adolescents âgés de 12 ans et plus.
La sécurité et l'efficacité du sémaglutide chez les enfants âgés de moins de 12 ans n'ont pas été établies.
Mode d'administration
Voie sous-cutanée.
Wegovy doit être administré une fois par semaine, quel que soit le moment de la journée, au cours ou en dehors des repas.
Il doit être injecté par voie sous-cutanée dans l'abdomen, la cuisse ou le haut du bras. Le site d'injection peut être modifié sans ajustement de la dose. Il ne doit pas être administré par voie intraveineuse ou intramusculaire.
Le jour de l'administration hebdomadaire peut être changé si nécessaire, à condition que le délai entre deux doses soit d'au moins 3 jours (> 72 heures). Après avoir choisi un nouveau jour d'administration, il faut continuer d'administrer la dose une fois par semaine.
Lors de l'administration de Wegovy stylo prérempli à dose unique, le stylo doit être appuyé fermement contre la peau jusqu'à ce que la barre jaune se soit immobilisée. L'injection dure environ 5 à 10 secondes.
Il convient de conseiller aux patients de lire attentivement les instructions d'utilisation incluses dans la notice avant l'administration du médicament.
Pour les instructions plus détaillées avant l'administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
Avant utilisation : 3 ans.
Après la première utilisation : 6 semaines. À conserver à une
température ne dépassant pas 30 °C ou au réfrigérateur (entre 2 °C et 8
°C).
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C). Maintenir à distance de l'élément de refroidissement. Ne pas congeler.
Laisser le capuchon du stylo en place lorsque le stylo n'est pas utilisé afin de le protéger de la lumière.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Un surdosage par le sémaglutide peut être associé à des troubles gastro-intestinaux qui pourraient conduire à une déshydratation. En cas de surdosage, un traitement symptomatique approprié doit être initié en fonction des signes cliniques observés chez le patient.
Classe pharmacothérapeutique : médicaments utilisés dans le diabète, analogue du glucagon-like peptide 1 (GLP-1), code ATC : A10BJ06
Mécanisme d'action
Le sémaglutide est un analogue du GLP-1 présentant 94 % d'homologie de séquence avec le GLP-1 humain. Le sémaglutide agit comme agoniste des récepteurs du GLP-1, qui se lie sélectivement et active le récepteur du GLP-1, la cible du GLP-1 natif.
Le GLP-1 est un régulateur physiologique de l'appétit et de l'apport calorique, et le récepteur du GLP- 1 est présent dans plusieurs régions du cerveau impliquées dans la régulation de l'appétit.
Les études chez l'animal montrent que le sémaglutide agit dans le cerveau par le biais du récepteur du GLP-1. Le sémaglutide exerce des effets directs sur les régions du cerveau impliquées dans la régulation de l'homéostasie de l'apport alimentaire dans l'hypothalamus et le tronc cérébral. Le sémaglutide peut agir sur le système de récompense hédonique à travers des effets directs et indirects dans des régions du cerveau, notamment le septum, le thalamus et l'amygdale.
Des études cliniques montrent que le sémaglutide réduit l'apport énergétique, augmente la sensation de satiété, de rassasiement et le contrôle de la prise alimentaire, réduit la sensation de faim, et la fréquence et l'intensité des fringales. En outre, le sémaglutide réduit la préférence pour les aliments à forte teneur en graisse.
Le sémaglutide régit les contributions homéostatique et hédonique en lien avec la fonction exécutive pour réguler l'apport calorique, l'appétit, la récompense et le choix de nourriture.
En outre, il a été montré dans des études cliniques que le sémaglutide réduit la glycémie de manière dépendante du glucose en stimulant la sécrétion d'insuline et en réduisant la sécrétion de glucagon lorsque la glycémie est élevée. Le mécanisme de réduction de la glycémie entraîne également un léger retard de la vidange gastrique en début de phase postprandiale. Lors d'une hypoglycémie, le sémaglutide diminue la sécrétion d'insuline sans altérer la sécrétion du glucagon.
Les récepteurs du GLP-1 sont également exprimés dans le cœur, le système vasculaire, le système immunitaire et les reins. Dans les études cliniques, le sémaglutide a eu un effet bénéfique sur les lipides plasmatiques, a réduit la tension artérielle systolique et a réduit l'inflammation. En outre, les études chez l'animal ont montré que le sémaglutide atténue le développement de l'athérosclérose et présente une action anti-inflammatoire dans le système cardiovasculaire.
Le mécanisme d'action du sémaglutide dans la réduction du risque cardiovasculaire est probablement multifactoriel. Il semble être en partie lié aux effets de la perte de poids et à l'impact sur les facteurs de risque cardiovasculaire connus (la réduction de la tension artérielle, l'amélioration du profil lipidique et du métabolisme du glucose, ainsi que les effets anti-inflammatoires démontrés par la diminution de la CRPus (protéine C-réactive ultra-sensible)). Cependant, le mécanisme précis de réduction du risque cardiovasculaire n'a pas encore été élucidé.
Effets pharmacodynamiques
Appétit,
apport énergétique et choix des aliments
Le sémaglutide réduit l'appétit en renforçant la sensation de
rassasiement et de satiété, tout en diminuant la faim et la consommation
prospective de nourriture. Dans l'essai de phase 1, l'apport énergétique
pendant un repas à volonté était inférieur de 35 % sous sémaglutide
par rapport au placebo après 20 semaines de traitement. Cette constatation a
été étayée par un meilleur contrôle de l'alimentation, une diminution des
envies alimentaires et une préférence relativement réduite pour les aliments à
haute teneur en graisse. Les envies alimentaires ont été évaluées de manière
plus approfondie dans l'étude STEP 5 au moyen d'un questionnaire sur le
contrôle de l'alimentation (CoEQ). A la semaine 104,
la différence de traitement estimée, tant pour le contrôle des envies que pour
les envies d'aliments salés, était significativement en faveur du sémaglutide, alors qu'aucun effet clair n'a été observé
pour les envies d'aliments sucrés.
Lipides
à jeun et postprandiaux
Par
comparaison au placebo, le sémaglutide diminue les
concentrations des triglycérides et des lipoprotéines de très basse densité
(VLDL) à jeun de 12 % et de 21 %, respectivement. La réponse postprandiale en
termes de triglycérides et de VLDL après un repas très gras a été réduite de
> 40 %.
Efficacité et sécurité cliniques
L'efficacité et la sécurité d'emploi du sémaglutide pour la gestion du poids en association avec une réduction de l'apport calorique et une augmentation de l'activité physique ont été évaluées dans quatre essais de phase 3a randomisés de 68 semaines, en double aveugle, contrôlés par placebo (STEP 1 à 4). Au total, 4 684 patients adultes (2 652 randomisés pour recevoir le traitement par sémaglutide) ont été inclus dans ces essais. De plus, l'efficacité et la sécurité du sémaglutide sur deux ans par rapport au placebo ont été évaluées dans le cadre d'un essai de phase 3b randomisé, en double aveugle, contrôlé par placebo (STEP 5), incluant 304 patients (152 avec le traitement par sémaglutide).
Le traitement par sémaglutide a démontré une perte de poids supérieure, cliniquement significative et durable par rapport au placebo chez les patients souffrant d'obésité (IMC ≥ 30 kg/m2) ou de surpoids (IMC ≥ 27 kg/m2 à < 30 kg/m2) et présentant au moins une comorbidité liée au poids. En outre, dans l'ensemble des essais, une proportion plus élevée de patients a obtenu une perte de poids ≥ 5 %, ≥ 10 %, ≥ 15 % et ≥ 20 % sous sémaglutide par rapport au placebo. La réduction du poids corporel a été observée indépendamment de la présence de symptômes gastro-intestinaux tels que les nausées, les vomissements ou la diarrhée.
Le traitement par sémaglutide a également montré des améliorations statistiquement significatives du tour de taille, de la tension artérielle systolique et du fonctionnement physique par rapport au placebo.
L'efficacité a été démontrée, indépendamment de l'âge, du sexe, de l'origine ethnique, du poids corporel à l'inclusion, de l'IMC, de la présence d'un diabète de type 2 et du niveau de la fonction rénale. Des variations de l'efficacité étaient présentes dans tous les sous-groupes. Une perte de poids relativement plus élevée a été observée chez les femmes et chez les patients sans diabète de type 2, ainsi que chez les patients ayant un poids corporel plus faible à l'inclusion par rapport à ceux ayant un poids plus élevé.
STEP 1 : gestion du poids
Dans un essai mené double aveugle de 68 semaines, 1 961 patients souffrant d'obésité (IMC ≥ 30 kg/m2) ou de surpoids (IMC ≥ 27 kg/m2 à < 30 kg/m2) et présentant au moins une comorbidité liée au poids ont été randomisés pour recevoir le sémaglutide ou un placebo. Tous les patients ont suivi un régime alimentaire hypocalorique et ont augmenté leur activité physique pendant toute la durée de l'essai.
Une perte de poids est survenue précocement et s'est poursuivie tout au long de l'essai. À la fin du traitement (semaine 68), la perte de poids était supérieure et cliniquement significative par rapport au placebo (voir Tableau 4 et Figure 1). En outre, une proportion plus élevée de patients a obtenu une perte de poids ≥ 5 %, ≥ 10 %, ≥ 15 % et ≥ 20 % sous sémaglutide par rapport au placebo (voir Tableau 4). Parmi les patients atteints de prédiabète à l'inclusion, une plus grande proportion de patients présentait un statut glycémique normal à la fin du traitement par sémaglutide par rapport au placebo (84,1 % vs. 47,8 %).
Tableau 4 STEP 1 : résultats à la semaine 68
| Wegovy | Placebo | |
| Ensemble d'analyse complet (N) | 1 306 | 655 |
| Poids corporel | ||
| Inclusion (kg) | 105,4 | 105,2 |
| Variation (%) par rapport à l'inclusion1,2 | -14,9 | -2,4 |
|
Différence
(%) par rapport au placebo1 [IC à 95 %] |
-12,4 [-13,4 ; -11,5]* | - |
| Variation (kg) par rapport à l'inclusion | -15,3 | -2,6 |
|
Différence
(kg) par rapport au placebo1 [IC à 95 %] |
-12,7 [-13,7 : -11,7] | - |
|
Patients
(%) ayant obtenu une perte de poids ≥ 5 %3 |
83,5* | 31,1 |
|
Patients
(%) ayant obtenu une perte de poids ≥ 10 %3 |
66,1* | 12,0 |
|
Patients
(%) ayant obtenu une perte de poids ≥ 15 %3 |
47,9* | 4,8 |
| Tour de taille (cm) | ||
| Inclusion | 114,6 | 114,8 |
| Variation par rapport à l'inclusion1 | -13,5 | -4,1 |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-9,4 [-10,3 : -8,5]* | - |
| Tension artérielle systolique (mmHg) | ||
| Inclusion | 126 | 127 |
| Variation par rapport à l'inclusion1 | -6,2 | -1,1 |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-5,1 [-6,3 ; -3,9]* | - |
* p <
0,0001 (bilatéral non ajusté) pour la supériorité.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 17,1 % et 22,4 % de patients randomisés sur le sémaglutide 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -16,9 % et de -2,4 % pour le sémaglutide 2,4 mg et le placebo.
3 Estimation à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse principale.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 17,1 % et 22,4 % de patients randomisés sur le sémaglutide 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -16,9 % et de -2,4 % pour le sémaglutide 2,4 mg et le placebo.
3 Estimation à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse principale.
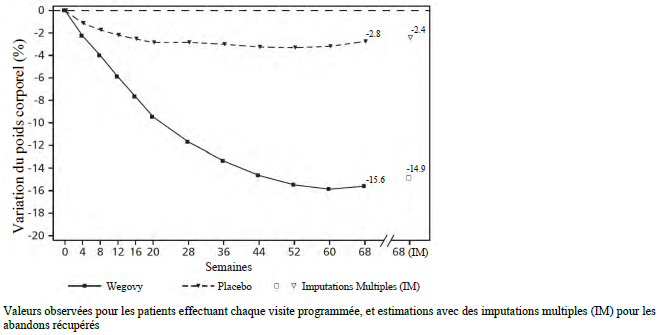
Figure 1 STEP 1 : variation moyenne du poids corporel (%) entre l'inclusion et la semaine 68
À la suite de l'essai de 68 semaines, une prolongation de 52 semaines sans traitement a été menée auprès de 327 patients qui avaient terminé la période principale de l'essai avec la dose d'entretien de sémaglutide ou le placebo. Au cours de la période sans traitement, de la semaine 68 à la semaine 120, le poids corporel moyen a augmenté dans les deux groupes de traitement. Cependant, chez les patients qui avaient été traités par sémaglutide pendant la période principale de l'essai, le poids est resté inférieur de 5,6 % à la valeur de départ, contre 0,1 % pour le groupe placebo.
STEP 2 :
gestion du poids chez les patients atteints de diabète de type 2
Dans une étude
menée en double aveugle de 68 semaines, 1 210 patients en surpoids ou souffrant
d'obésité (IMC ≥ 27 kg/m2) et atteints de diabète de type 2 ont
été randomisés pour recevoir du sémaglutide de 2,4
mg, du sémaglutide de 1 mg une fois par semaine ou un
placebo. Les patients inclus dans l'essai étaient atteints de diabète
insuffisamment contrôlé (HbA1c de 7 à 10 %) et traités par un régime
alimentaire et de l'activité physique uniquement ou par 1 à 3 antidiabétiques
oraux. Tous les patients ont suivi un régime alimentaire hypocalorique et ont
augmenté leur activité physique pendant toute la durée de l'essai.
Le traitement par sémaglutide pendant 68 semaines a entraîné une réduction supérieure et cliniquement significative du poids corporel et de l'HbA1c par rapport au placebo (voir Tableau 5 et Figure 2).
Tableau 5 STEP 2 : résultats à la semaine 68
| Wegovy | Placebo | |
| Ensemble d'analyse complet (N) | 404 | 403 |
| Poids corporel |
|
|
| Inclusion (kg) | 99,9 | 100,5 |
| Variation (%) par rapport à l'inclusion1,2 | -9,6 | -3,4 |
|
Différence
(%) par rapport au placebo1 [IC à 95 %] |
-6,2 [-7,3 ; -5,2]* | - |
| Variation (kg) par rapport à l'inclusion | -9,7 | -3,5 |
|
Différence
(kg) par rapport au placebo1 [IC à 95 %] |
-6,1 [7,2 ; -5,0] | - |
|
Patients
(%) ayant obtenu une perte de poids ≥ 5 %3 |
67,4* | 30,2 |
|
Patients
(%) ayant obtenu une perte de poids ≥ 10 %3 |
44,5* | 10,2 |
|
Patients
(%) ayant obtenu une perte de poids ≥ 15 %3 |
25,0* | 4,3 |
| Tour de taille (cm) | ||
| Inclusion | 114,5 | 115,5 |
| Variation par rapport à l'inclusion1 | -9,4 | -4,5 |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-4,9 [-6,0, -3,8]* | - |
| Tension artérielle systolique (mmHg) | ||
| Inclusion | 130 | 130 |
| Variation par rapport à l'inclusion1 | -3,9 | -0,5 |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-3,4 [-5,6 ; -1,3]** | - |
| HbA1c (%) (mmol/mol [%]) | ||
| Inclusion | 65,3 (8,1) | 65,3 (8,1) |
| Variation par rapport à l'inclusion1 | -17,5 (-1,6) | -4,1 (-0,4) |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-13,5
[-15,5 ; -11,4] (-1,2 [-1,4 ; -1,1])* |
- - |
* p <
0,0001 (bilatéral non ajusté) pour la supériorité ; **p < 0,05 (bilatéral
non ajusté) pour la supériorité.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 11,6 % et 13,9 % des patients randomisés sur le sémaglutide de 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations estimées entre la randomisation et la semaine 68 pour le poids corporel basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -10,6 % et de -3,1 % pour le sémaglutide de 2,4 mg et le placebo.
3 Estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse principale.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 11,6 % et 13,9 % des patients randomisés sur le sémaglutide de 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations estimées entre la randomisation et la semaine 68 pour le poids corporel basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -10,6 % et de -3,1 % pour le sémaglutide de 2,4 mg et le placebo.
3 Estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse principale.
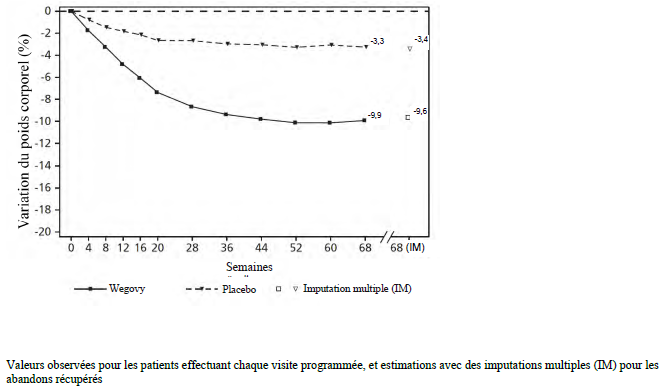
Figure 2 STEP 2 : variation moyenne du poids corporel (%) entre l'inclusion et la semaine 68
STEP 3 :
gestion du poids par une thérapie comportementale intensive
Dans un essai
mené en double aveugle de 68 semaines, 611 patients souffrant d'obésité (IMC ≥
30 kg/m2) ou de surpoids (IMC ≥ 27 kg/m2 à < 30
kg/m2) et présentant au moins une comorbidité liée au poids ont été
randomisés pour recevoir le sémaglutide ou un
placebo. Pendant l'essai, tous les patients ont bénéficié d'une thérapie comportementale
intensive (IBT) consistant en un régime alimentaire très restrictif, une
augmentation de l'activité physique et des conseils comportementaux.
Le traitement par sémaglutide et IBT pendant 68 semaines a entraîné une réduction du poids corporel supérieure et cliniquement significative par rapport au placebo (voir Tableau 6).
Tableau 6 STEP 3 : résultats à la semaine 68
| Wegovy | Placebo | |
| Ensemble d'analyse complet (N) | 407 | 204 |
| Poids corporel | ||
| Inclusion (kg) | 106,9 | 103,7 |
| Variation (%) par rapport à l'inclusion1,2 | -16,0 | -5,7 |
|
Différence
(%) par rapport au placebo1 [IC à 95 %] |
-10,3 [-12,0 ; -8,6]* | - |
| Variation (kg) par rapport à l'inclusion | -16,8 | -6,2 |
|
Différence
(kg) par rapport au placebo1 [IC à 95 %] |
-10,6 [-12,5 ; -8,8] | - |
|
Patients
(%) ayant obtenu une perte de poids ≥ 5 %3 |
84,8* | 47,8 |
|
Patients
(%) ayant obtenu une perte de poids ≥ 10 %3 |
73,0* | 27,1 |
|
Patients
(%) ayant obtenu une perte de poids ≥ 15 %3 |
53,5* | 13,2 |
| Tour de taille (cm) | ||
| Inclusion | 113,6 | 111,8 |
| Variation par rapport à l'inclusion1 | -14,6 | -6,3 |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-8,3 [-10,1 ; -6,6]* | - |
| Tension artérielle systolique (mmHg) | ||
| Inclusion | 124 | 124 |
| Variation par rapport à l'inclusion1 | -5,6 | -1,6 |
|
Différence
par rapport au placebo1 [IC à 95 %] |
-3,9 [-6,4 ; -1,5]* | - |
* p < 0,005
(bilatéral non ajusté) pour la supériorité.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 16,7 % et 18,6 % des patients randomisés pour recevoir le sémaglutide de 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -17,6 % et de -5,0 % pour le sémaglutide de 2,4 mg et le placebo. 3 Estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse principale.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 16,7 % et 18,6 % des patients randomisés pour recevoir le sémaglutide de 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -17,6 % et de -5,0 % pour le sémaglutide de 2,4 mg et le placebo. 3 Estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse principale.
STEP 4 :
gestion durable du poids
Dans un essai
mené en double aveugle de 68 semaines, 902 patients souffrant d'obésité (IMC ≥
30 kg/m2) ou de surpoids (IMC ≥ 27 kg/m2 à < 30
kg/m2) et présentant au moins une comorbidité liée au poids ont été
inclus. Tous les patients ont suivi un régime alimentaire hypocalorique et ont
augmenté leur activité physique pendant toute la durée de l'essai. Entre la
semaine 0 et la semaine 20 (pré-inclusion), tous les patients ont reçu du sémaglutide. À la semaine 20 (inclusion), les patients qui
avaient atteint la dose d'entretien de 2,4 mg ont été randomisés pour
poursuivre le traitement ou utiliser le placebo. À la semaine 0 (début de la
période de pré-inclusion), le poids corporel moyen des patients était de 107,2
kg et l'IMC moyen de 38,4 kg/m2.
Les patients qui avaient atteint la dose d'entretien de 2,4 mg à la semaine 20 (inclusion) et qui ont poursuivi le traitement par sémaglutide pendant 48 semaines (semaines 20 à 68) ont continué à perdre du poids et ont obtenu une réduction de poids corporel supérieure et cliniquement significative par rapport à ceux sous placebo (voir Tableau 7 et Figure 3). Le poids corporel a augmenté régulièrement entre la semaine 20 et la semaine 68 chez les patients sous placebo depuis la semaine 20 (inclusion). Néanmoins, le poids corporel moyen observé était inférieur à la semaine 68 par rapport au début de la période de pré-inclusion (semaine 0) (voir Figure 3). Les patients traités par sémaglutide entre la semaine 0 (pré-inclusion) et la semaine 68 (fin du traitement) ont obtenu une variation moyenne de poids corporel de 17,4 %, avec une perte de poids ≥ 5 % obtenue par 87,8 %, ≥ 10 % par 78,0 %, ≥ 15 % par 62,2 % et ≥ 20 % par 38,6 % de ces patients.
Tableau 7 STEP 4 : Résultats entre la semaine
20 et la semaine 68
| Wegovy | Placebo | |
| Ensemble d'analyse complet (N) | 535 | 268 |
| Poids corporel | ||
| Inclusion1 (kg) | 96,5 | 95,4 |
| Variation (%) par rapport à l'inclusion1,2,3 | -7,9 | 6,9 |
|
Différence
(%) par rapport au placebo2 [IC à 95 %] |
-14,8 [-16,0 ; -13,5]* | - |
| Variation (kg) par rapport à l'inclusion | -7,1 | 6,1 |
|
Différence
(kg) par rapport au placebo2 [IC à 95 %] |
-13,2 [-14,3 ; -12,0] | - |
| Tour de taille (cm) | ||
| Inclusion | 105,5 | 104,7 |
| Variation par rapport à l'inclusion1 | -6,4 | 3,3 |
| Différence
par rapport au placebo2 [IC à 95 %] |
-9,7 [-10,9 ; -8,5]* | - |
| Tension artérielle systolique (mmHg) | ||
| Inclusion1 | 121 | 121 |
| Variation par rapport à l'inclusion1,2 | 0,5 | 4,4 |
|
Différence
par rapport au placebo2 [IC à 95 %] |
-3,9 [-5,8 ; -2,0]* | - |
* p <
0,0001 (bilatéral non ajusté) pour la supériorité.
1 Inclusion = semaine 20
2 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
3 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 5,8 % et 11,6 % des patients randomisés pour recevoir le sémaglutide de 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -8,1 % et de 6,5 % pour le sémaglutide de 2,4 mg et le placebo.
1 Inclusion = semaine 20
2 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
3 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 5,8 % et 11,6 % des patients randomisés pour recevoir le sémaglutide de 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -8,1 % et de 6,5 % pour le sémaglutide de 2,4 mg et le placebo.
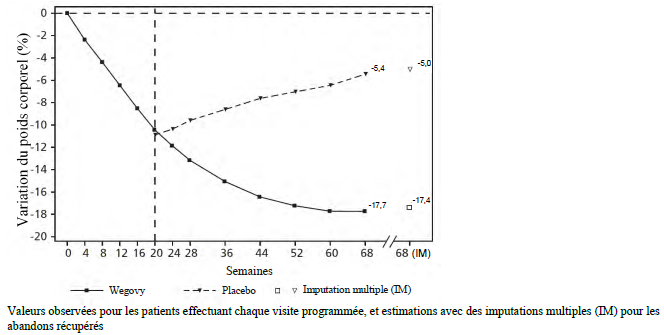
Figure 3 STEP 4 : variation moyenne du poids corporel (%) entre la semaine 0 et la semaine 68
STEP 5 :
Données sur 2 ans
Dans un essai
en double aveugle de 104 semaines, 304 patients souffrant d'obésité (IMC ≥
30 kg/m²), ou de surpoids (IMC ≥ 27 kg/m² à < 30 kg/m²) et présentant
au moins une comorbidité liée au poids, ont été randomisés entre le sémaglutide et le placebo. Tous les patients ont suivi un
régime hypocalorique et ont augmenté leur activité physique pendant toute la
durée de l'essai. A l'inclusion, les patients avaient un IMC moyen de 38,5
kg/m² et un poids corporel moyen de 106,0 kg.
Le traitement par sémaglutide pendant 104 semaines a entraîné une réduction supérieure et cliniquement significative du poids corporel par rapport au placebo. Le poids corporel moyen a diminué, de l'inclusion à la semaine 68 avec le sémaglutide, suivi de l'atteinte d'un plateau. Avec le placebo, le poids corporel moyen a moins diminué, et un plateau a été atteint après environ 20 semaines de traitement (voir tableau 8 et figure 4). Les patients traités par sémaglutide ont atteint une variation moyenne du poids corporel de -15,2 %, avec une perte de poids ≥ 5 % atteinte par 74,7 %, ≥ 10 % atteinte par 59,2 % et ≥ 15% atteinte par 49,7 % de ces patients. Parmi les patients présentant un prédiabète au départ, 80 % et 37 % ont atteint un statut normo-glycémique à la fin du traitement par sémaglutide et le placebo, respectivement.
Tableau 8 STEP 5 : Résultats à la semaine 104
| Wegovy | Placebo | |
| Ensemble d'analyse complet (N) | 152 | 152 |
| Poids corporel | ||
| Inclusion1 (kg) | 105,6 | 106,5 |
| Variation (%) par rapport à l'inclusion1,2,3 | -15,2 | -2,6 |
|
Différence
(%) par rapport au placebo1 [IC à 95 %] |
-12,6 [-15,3 ; -9,8]* | - |
| Variation (kg) par rapport à l'inclusion | -16,1 | -3,2 |
|
Différence
(kg) par rapport au placebo2 [IC à 95 %] |
-12,9 [-16,1 ; -9,8] | - |
| Patients (%) avec une perte de poids > 5 %3 | 74,7* | 37,3 |
| Patients (%) avec une perte de poids > 10 %3 | 59,2* | 16,8 |
| Patients (%) avec une perte de poids > 15 %3 | 49,7* | 9,2 |
| Tour de taille (cm) | ||
| Inclusion | 115,8 | 115,7 |
| Variation par rapport à l'inclusion1 | -14,4 | 5,2 |
|
Différence
par rapport au placebo2 [IC à 95 %] |
-9,2 [-12,2 ; -6,2]* | - |
| Tension artérielle systolique (mmHg) | ||
| Inclusion1 | 126 | 125 |
| Variation par rapport à l'inclusion1,2 | -5,7 | -1,6 |
|
Différence
par rapport au placebo2 [IC à 95 %] |
-4,2 [-7,3 ; -1,0]* |
|
* p <
0,0001 (bilatéral non ajusté) pour la supériorité
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 13,2 % et 27,0 % des patients randomisés pour recevoir le sémaglutide et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -16,7 % et de -0,6 % pour le sémaglutide et le placebo.
3 Valeur estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse primaire.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 13,2 % et 27,0 % des patients randomisés pour recevoir le sémaglutide et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -16,7 % et de -0,6 % pour le sémaglutide et le placebo.
3 Valeur estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse primaire.
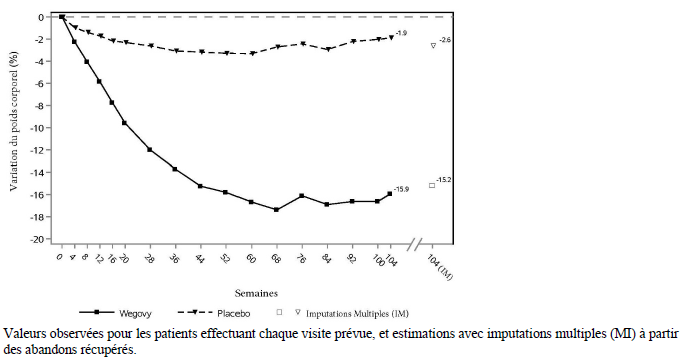
Figure 4 STEP 5 : Changement moyen du poids corporel (%) de la semaine 0 à la semaine 104
STEP 8 : Sémaglutide vs liraglutide
Dans un essai
de 68 semaines, randomisé, ouvert, contrôlé par placebo, 338 patients souffrant
d'obésité (IMC ≥ 30 kg/m²), ou de surpoids (IMC ≥ 27 kg/m² à <
30 kg/m²) et d'au moins une comorbidité liée au poids, ont été randomisés entre
le sémaglutide une fois par semaine, le liraglutide 3 mg une fois par jour ou le placebo. Le sémaglutide une fois par semaine et le liraglutide
3 mg ont été évalués en ouvert, mais chaque groupe de traitement actif a été
évalué en double aveugle par rapport au placebo administré à la même fréquence.
Tous les patients ont suivi un régime hypocalorique et ont augmenté leur
activité physique pendant toute la durée de l'essai. A l'inclusion, les
patients avaient un IMC moyen de 37,5 kg/m² et un poids corporel moyen de 104,5
kg.
Le traitement par sémaglutide une fois par semaine pendant 68 semaines a entraîné une réduction supérieure et cliniquement significative du poids corporel par rapport au liraglutide. Le poids corporel moyen a diminué, de l'inclusion à la semaine 68 avec le sémaglutide. Avec le liraglutide le poids corporel moyen a moins diminué (voir tableau 9). 37,4 % des patients traités par sémaglutide ont perdu ≥ 20 %, contre 7,0 % traités par le liraglutide. Le tableau 9 présente les résultats des critères de confirmation ≥ 10 %, ≥ 15 % et ≥ 20 % de perte de poids.
Tableau 9 STEP 8 : Résultats d'un essai
clinique de 68 semaines comparant le sémaglutide au liraglutide
| Wegovy | Liraglutide 3 mg | |
| Ensemble d'analyse complet (N) | 126 | 127 |
| Poids corporel | ||
| Inclusion1 (kg) | 102,5 | 103,7 |
| Variation (%) par rapport à l'inclusion1,2,3 | -15,8 | -6,4 |
|
Différence
(%) par rapport au liraglutide1 [IC à 95 %] |
-9,4 [-12,0 ; -6,8]* | - |
| Variation (kg) par rapport à l'inclusion | -15,3 | -6,8 |
|
Différence
(kg) par rapport au liraglutide2 [IC à 95 %] |
-8,5 [-11,2 ; -5,7] | - |
| Patients (%) avec une perte de poids > 10 %3 | 69,4* | 27,2 |
| Patients (%) avec une perte de poids > 15 %3 | 54,0* | 13,4 |
| Patients (%) avec une perte de poids > 20 %3 | 37,4* | 7,0 |
* p < 0,0001
(bilatéral non ajusté) pour la supériorité.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 13,5 % et 27,6 % des patients randomisés pour recevoir le sémaglutide et le liragludite. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -16,7 % et de -6,7 % pour le sémaglutide et le liraglutide.
3 Valeur estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse primaire.
1 Valeur estimée au moyen d'un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l'arrêt du traitement randomisé ou de l'instauration d'un autre médicament contre l'obésité ou d'une chirurgie bariatrique.
2 Pendant l'essai, le traitement randomisé a été définitivement interrompu par respectivement 13,5 % et 27,6 % des patients randomisés pour recevoir le sémaglutide et le liragludite. En supposant que tous les patients randomisés sont restés sous traitement et n'ont pas reçu d'autres médicaments contre l'obésité, les variations de poids corporel estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu'au premier arrêt étaient respectivement de -16,7 % et de -6,7 % pour le sémaglutide et le liraglutide.
3 Valeur estimé à partir d'un modèle de régression binaire basé sur la même procédure d'imputation que dans l'analyse primaire.
Effet sur
la composition corporelle
Dans une
sous-étude de STEP 1 (N = 140), la composition corporelle a été mesurée par absorptiométriebiphotonique à
rayons X (DEXA). Les résultats de l'évaluation DEXA a montré que le traitement
par sémaglutide s'accompagne d'une plus grande
réduction de masse grasse que de masse corporelle maigre, ce qui entraîne une
amélioration de la composition corporelle par rapport au placebo après 68
semaines. En outre, cette réduction de masse grasse totale s'accompagnait d'une
réduction de la graisse viscérale. Ces résultats suggèrent que la plupart de la
perte de poids totale était attribuable à une réduction du tissu adipeux,
notamment de la graisse viscérale.
Amélioration
du fonctionnement physique
Le sémaglutide a entrainé des légères améliorations
statistiquement significatives des scores de fonctionnement physique. Le
fonctionnement physique a été évalué par le questionnaire générique sur la
qualité de vie liée à la santé de l'enquête de santé abrégée à 36 items, v2,
version aiguë (SF-36) et par le questionnaire spécifique à l'obésité sur
l'impact du poids sur la qualité de vie, version allégée pour les essais
cliniques (IWQOL-Lite-CT).
Évaluation cardiovasculaire
SELECT : Essai de morbi-mortalité cardiovasculaire chez des patients en situation de surpoids ou d'obésité.
SELECT était
un essai piloté par les événements, randomisé, en double aveugle, contrôlé par
placebo, ayant inclus 17 604 patients présentant une maladie cardiovasculaire
établie et un IMC ≥ 27 kg/m2.
Les patients
ont été randomisés pour recevoir le sémaglutide 2,4
mg (n = 8 803) ou un placebo (n = 8 801) en complément d'une prise en charge
standard. La durée médiane de participation à l'essai était de 41,8 mois. Le
statut vital était disponible pour 99,4 % des patients de l'essai.
La population étudiée était composée de 27,7 % de femmes et de 72,3 % d'hommes, avec une moyenne d'âge de 61,6 ans, dont 38,2 % de patients âgés de ≥ 65 ans (n = 6 728) et 7,8 % de patients âgés de ≥ 75 ans (n = 1 366). L'IMC moyen était de 33,3 kg/m2 et le poids corporel moyen était de 96,7 kg. Les patients ayant des antécédents de diabète de type 1 et de type 2 étaient exclus.
Le critère d'évaluation principal était le délai de survenue du premier événement cardiovasculaire majeur (MACE) depuis la randomisation, défini comme un critère composite regroupant la mortalité cardiovasculaire (y compris la cause de décès indéterminée), l'infarctus du myocarde non fatal ou l'accident vasculaire cérébral non fatal. Le critère principal, le temps jusqu'au premier MACE, s'est produit chez 1 270 des 17 604 patients inclus dans l'essai SELECT. Plus précisément, 569 premiers MACE (6,5 %) ont été enregistrés parmi les 8 803 patients traités par sémaglutide, comparé à 701 premiers MACE (8,0 %) parmi les 8 801 patients traités par placebo. Un total de 63 (11,1 %) des premiers MACE avec le sémaglutide et 80 (11,4 %) avec le placebo étaient dus à une cause de décès indéterminée.
La supériorité du sémaglutide 2,4 mg par rapport au placebo concernant les MACE a été confirmée avec un risque relatif de 0,80 [0,72 ; 0,90] [IC à 95 %] correspondant à une réduction du risque relatif de MACE de 20 % (voir Figure 5). La contribution de chaque composant du critère composite à la réduction des MACE est présentée dans la Figure 6. La réduction des MACE avec le sémaglutide 2,4 mg n'a pas été affectée par l'âge, le sexe, l'origine ethnique, l'IMC à l'inclusion ou le niveau d'insuffisance rénale.
L'analyse de la mortalité cardiovasculaire (premier critère d'évaluation secondaire de confirmation) a généré un risque relatif de 0,85 [0,71 ; 1,01] [IC à 95 %].
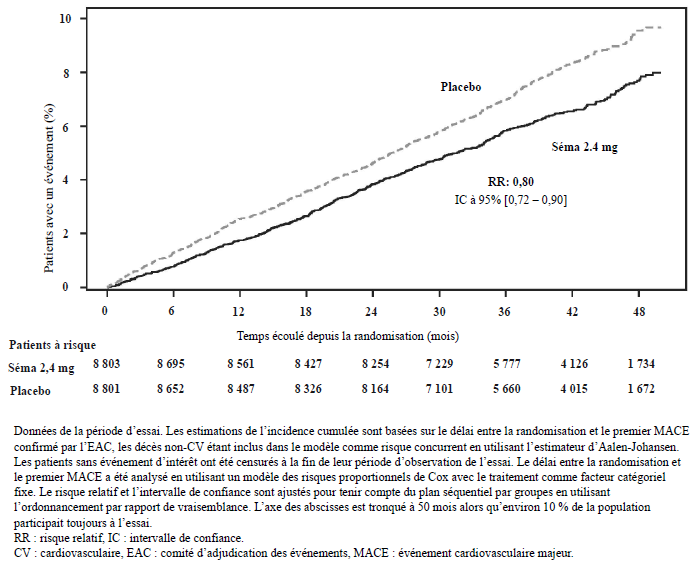
Figure 5 : Graphique de la fonction d'incidence cumulée du délai entre la randomisation et la survenue du premier MACE

Figure 6 : Graphique en forêt du délai entre la randomisation et la survenue du premier MACE, des composants du critère composite des MACE et des critères d'évaluation secondaires de confirmation
SUSTAIN 6 : Essai de morbi-mortalité cardiovasculaire chez des patients atteints de diabète de type 2
Dans l'étude SUSTAIN 6, 3 297 patients atteints diabète de type 2 insuffisamment contrôlé présentant un risque élevé d'événements cardiovasculaires ont été randomisés pour recevoir le sémaglutide sous cutané 0,5 mg ou 1 mg une fois par semaine ou le placebo en plus de la prise en charge standard. La durée du traitement était de 104 semaines. L'âge moyen était de 65 ans et l'IMC moyen de 33 kg/m2.
Le critère d'évaluation principal était le délai de survenue, depuis la randomisation, du premier événement cardiovasculaire majeur (MACE) : mortalité cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal. Le nombre total de MACE était de 254, dont 108 (6,6 %) avec le sémaglutide et 146 (8,9 %) avec un placebo.
La sécurité cardiovasculaire du traitement par sémaglutide 0,5 ou 1 mg a été confirmée, car le risque relatif (RR) sous sémaglutide par rapport au placebo était de 0,74, [0,58, 0,95] [IC à 95 %], grâce à une diminution du taux d'accident vasculaire cérébral non fatal et d'infarctus du myocarde non fatal sans différence au niveau des décès d'origine cardiovasculaire (voir Figure 7).
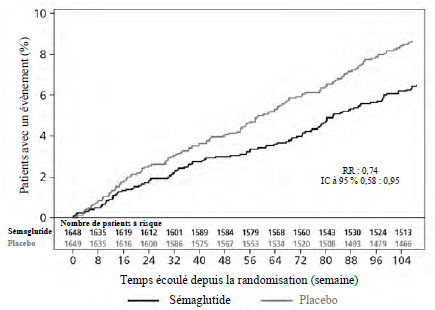
Figure 7 : représentation Kaplan-Maier du délai de survenue du premier événement composite : mortalité cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal (SUSTAIN 6)
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Wegovy dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de la gestion du poids (voir rubrique 4.2 pour les informations concernant l'usage pédiatrique).
STEP TEENS : Gestion du poids chez les patients adolescents
Dans un essai mené en double aveugle de 68 semaines, 201 adolescents pubères âgés de 12 à < 18 ans, souffrant d'obésité ou de surpoids et présentant au moins une comorbidité liée au poids ont été randomisés selon un rapport de 2:1 pour recevoir le sémaglutide ou un placebo. Tous les patients ont suivi un régime hypocalorique et ont augmenté leur activité physique pendant toute la durée de l'essai.
À la fin du traitement (semaine 68), l'amélioration de l'IMC avec le sémaglutide était supérieure et cliniquement significative par rapport au placebo (voir Tableau 10 et Figure 8). En outre, une proportion plus élevée de patients a obtenu une perte de poids ≥ 5 %, ≥ 10 % et ≥ 15 % sous sémaglutide par rapport au placebo (voir Tableau 10).
Tableau 10
STEP TEENS : Résultats à la semaine 68
| Wegovy | Placebo | |
| Ensemble d'analyse complet (N) | 134 | 67 |
| IMC | ||
| Inclusion (IMC) | 37,7 | 35,7 |
| Variation (%) par rapport à l'inclusion1,2, | -16,1 | 0,6 |
| Différence (%) par rapport au placebo1 [IC à 95 %] | -16,7 [-20,3 ; -13,2]* | - |
| Inclusion (IMC SDS) | 3,4 | 3,1 |
| Variation par rapport à l'inclusion de l'IMC SDS1 | -1,1 | -0 1 |
| Différence par rapport au placebo1 [IC à 95 %] | -1,0 [-1,3 ; -0,8] | - |
| Poids corporel |
|
|
| Inclusion (kg) | 109,9 | 102,6 |
| Variation (%) par rapport à l'inclusion1 | -14,7 | 2,8 |
| Différence (%) par rapport au placebo1 [IC à 95 %] | -17,4 [-21,1 ; -13,8] | - |
| Variation (kg) par rapport à l'inclusion1 | -15,3 | 2,4 |
| Différence (kg) par rapport au placebo1 [IC à 95 %] | -17,7 [-21,8 ; -13,7] | - |
| Patients (%) avec une perte de poids ≥ 5 %3 | 72,5* | 17,7 |
| Patients (%) avec une perte de poids ≥ 10 %3 | 61,8 | 8,1 |
| Patients (%) avec une perte de poids ≥ 15 %3 | 53,4 | 4,8 |
| Tour de taille (cm) | ||
| Inclusion | 111,9 | 107,3 |
| Variation par rapport à l'inclusion1 | -12,7 | -0,6 |
| Différence par rapport au placebo1 [IC à 95 %] | -12,1 [-15,6 ; -8,7] | - |
| Tension artérielle systolique (mmHg) | ||
| Inclusion | 120 | 120 |
| Variation par rapport à l'inclusion1 | -2,7 | -0,8 |
| Différence par rapport au placebo1 [IC à 95 %] | -1,9 [-5,0 ; 1,1] | - |
* p < 0,0001 (bilatéral non ajusté) pour la supériorité.
1 Valeur estimée au moyen d’un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l’arrêt du traitement randomisé ou de l’instauration d’un autre médicament contre l’obésité ou d’une chirurgie bariatrique.
2 Pendant l’essai, le traitement randomisé a été définitivement interrompu par respectivement 10,4 % et 10,4 % des patients randomisés pour recevoir le sémaglutide 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n’ont pas reçu d’autres médicaments contre l’obésité, les variations de l’IMC estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu’au premier arrêt étaient respectivement de -17,9 % et de 0,6 % pour le sémaglutide 2,4 mg et le placebo.
3 Valeur estimée à partir d’un modèle de régression binaire basé sur la même procédure d’imputation que dans l’analyse primaire.
1 Valeur estimée au moyen d’un modèle ANCOVA utilisant une imputation multiple basée sur toutes les données indépendamment de l’arrêt du traitement randomisé ou de l’instauration d’un autre médicament contre l’obésité ou d’une chirurgie bariatrique.
2 Pendant l’essai, le traitement randomisé a été définitivement interrompu par respectivement 10,4 % et 10,4 % des patients randomisés pour recevoir le sémaglutide 2,4 mg et le placebo. En supposant que tous les patients randomisés sont restés sous traitement et n’ont pas reçu d’autres médicaments contre l’obésité, les variations de l’IMC estimées entre la randomisation et la semaine 68 basées sur un modèle mixte pour mesures répétées comprenant toutes les observations jusqu’au premier arrêt étaient respectivement de -17,9 % et de 0,6 % pour le sémaglutide 2,4 mg et le placebo.
3 Valeur estimée à partir d’un modèle de régression binaire basé sur la même procédure d’imputation que dans l’analyse primaire.
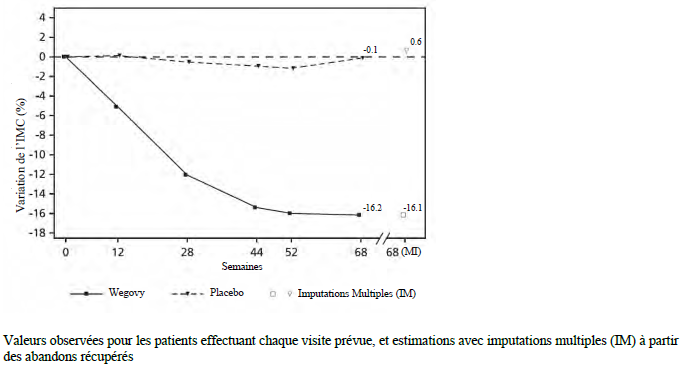
Figure 8 STEP TEENS : Variation moyenne de l'IMC (%) de l'inclusion à la semaine 68
En comparaison avec le GLP-1 natif, le sémaglutide a une demi-vie prolongée d'environ une semaine, ce qui permet de l'administrer par voie sous-cutanée une fois par semaine. Le principal mécanisme d'action prolongée est la liaison à l'albumine, qui entraîne une baisse de la clairance rénale et une protection contre la dégradation métabolique. De plus, le sémaglutide est stabilisé de manière à éviter la dégradation par l'enzyme DPP-4.
Absorption
La concentration moyenne de sémaglutide à l'état d'équilibre après une administration sous-cutanée de la dose d'entretien de sémaglutide était environ 75 nmol/l chez les patients en surpoids (IMC≥ 27 kg/m2 à < 30 kg/m2) ou souffrant d'obésité (IMC ≥ 30 kg/m2) d'après des données issues d'études de phase 3a, dans lesquelles 90 % des patients avaient des concentrations moyennes comprises entre 51 nmol/l et 110 nmol/l. L'exposition au sémaglutide à l'état d'équilibre a augmenté proportionnellement à la dose entre 0,25 mg et 2,4 mg une fois par semaine. L'exposition à l'état d'équilibre était stable au fil du temps, et a été évaluée jusqu'à la semaine 68. Une exposition similaire a été obtenue avec une administration sous-cutanée de sémaglutide dans l'abdomen, la cuisse ou le haut du bras. La biodisponibilité absolue du sémaglutide était de 89 %.
Distribution
Le volume de distribution moyen du sémaglutide après administration sous-cutanée chez des patients en surpoids ou souffrant d'obésité était d'environ 12,4 l. Le sémaglutide était fortement lié à l'albumine plasmatique (> 99 %).
Métabolisme/biotransformation
Avant l'excrétion, le sémaglutide est fortement métabolisé par clivage protéolytique de la chaîne peptidique et bêta-oxydation séquentielle de la chaîne latérale d'acides gras. L'enzyme endopeptidase neutre (EPN) interviendrait dans le métabolisme du sémaglutide.
Élimination
Les
principales voies d'excrétion des substances apparentées au sémaglutide
sont l'urine et les fèces. Approximativement 3 % de la dose absorbée
est excrétée sous la forme de sémaglutide intact dans l'urine.
La clairance du sémaglutide chez les patients en surpoids (IMC ≥ 27 kg/m2 à < 30 kg/m2) ou souffrant d'obésité (IMC ≥ 30 kg/m2)
était environ 0,05 l/heure. Avec une demi-vie d'élimination d'environ 1
semaine, le sémaglutide restera présent dans la circulation pendant
approximativement 7 semaines après la dernière dose de 2,4 mg.
Populations particulières
Sujets âgés
L'âge
n'a aucun effet sur la pharmacocinétique du sémaglutide, selon les
données des études de phase 3 portant sur des patients âgés de 18 à 86
ans.
Genre et origine ethnique
Le
genre et l'origine ethnique (Blanc, Noir, Afro-Américain, Asiatique ;
Hispanique ou Latino, non-Hispanique ou non-Latino) n'ont eu aucun
effet sur la pharmacocinétique du sémaglutide, selon les données des
études de phase 3a.
Poids corporel
Le
poids corporel influence l'exposition au sémaglutide. Un poids corporel
plus élevé diminue l'exposition ; une différence de 20 % de poids
corporel entre les patients entraîne une différence d'environ 18 % de
l'exposition. La dose hebdomadaire de 2,4 mg de sémaglutide assure des
expositions systémiques adéquates à un poids corporel dans l'intervalle
compris entre 54,4 et 245,6 kg, évalué pour la réponse à l'exposition
dans les essais cliniques.
Insuffisance rénale
L'insuffisance
rénale n'a pas affecté la pharmacocinétique du sémaglutide de manière
cliniquement significative. Cela a été observé avec une dose unique de
0,5 mg de sémaglutide chez des patients présentant des degrés divers
d'insuffisance rénale (légère, modérée, sévère ou patients dialysés) en
comparaison avec des patients à la fonction rénale normale. Cela a
également été observé chez les patients en surpoids (IMC ≥ 27 kg/m 2 à < 30 kg/m2) ou souffrant d'obésité (IMC ≥ 30 kg/m2) et présentant une insuffisance rénale légère à modérée d'après les données des essais de phase 3a.
Insuffisance hépatique
L'insuffisance
hépatique n'a aucun impact sur l'exposition au sémaglutide. La
pharmacocinétique du sémaglutide a été évaluée chez des patients
présentant des degrés divers d'insuffisance hépatique (légère, modérée,
sévère) en comparaison avec des patients présentant une fonction
hépatique normale dans le cadre d'une étude utilisant une dose unique
de 0,5 mg de sémaglutide.
Prédiabète et diabète
Le
prédiabète et le diabète n'ont eu aucun effet cliniquement pertinent
sur l'exposition au sémaglutide, d'après les données des essais de
phase 3.
Immunogénicité
Le
développement d'anticorps anti-sémaglutide suite à un traitement par
sémaglutide a été observé peu fréquemment (voir rubrique Effets indésirables) et la réponse ne semblait pas influencer la pharmacocinétique du sémaglutide.
Population pédiatrique
Les
propriétés pharmacocinétiques du sémaglutide ont été évaluées dans un
essai clinique chez des patients adolescents souffrant d'obésité ou de
surpoids et présentant au moins une comorbidité liée au poids, âgés de
12 à < 18 ans (124 patients, poids corporel compris entre 61,6 et
211,9 kg).
L'exposition au sémaglutide chez les adolescents a été similaire à celle chez les adultes souffrant d'obésité ou de surpoids.
La sécurité et l'efficacité du sémaglutide chez les enfants âgés de moins de 12 ans n'ont pas été étudiées.
Le sémaglutide n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, des vertiges peuvent être ressentis principalement durant la période d'augmentation de la dose. La conduite de véhicules ou l'utilisation de machines doivent être effectuées avec prudence en cas de vertiges.
Patients ayant un diabète de type 2
Lorsque le sémaglutide est utilisé en association à un sulfamide hypoglycémiant ou à l'insuline, il convient d'informer les patients qu'ils doivent prendre des précautions pour éviter une hypoglycémie lors de la conduite de véhicules ou de l'utilisation de machines (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée ou génotoxicité n'ont pas révélé de risque particulier pour l'homme.
Les tumeurs non létales des cellules C de la thyroïde observées chez les rongeurs constituent un effet spécifique à la classe des agonistes des récepteurs du GLP-1. Lors d'études de carcinogénicité sur 2 ans chez le rat et la souris, le sémaglutide a provoqué des tumeurs des cellules C de la thyroïde à des expositions cliniquement significatives. Aucun autre type de tumeurs liées au traitement n'a été observé. Les tumeurs des cellules C chez les rongeurs sont dues à un mécanisme non génotoxique, spécifique, médié par les récepteurs du GLP-1, auquel les rongeurs sont particulièrement sensibles. La pertinence de ces résultats pour l'homme est considérée comme faible, mais ne peut pas être complètement exclue.
Lors d'études de fertilité chez le rat, le sémaglutide n'a pas affecté les performances d'accouplement ni la fertilité des mâles. Chez le rat femelle, une prolongation du cycle œstrien et une faible baisse du nombre d'ovulations ont été observées à des doses associées à une réduction du poids maternel.
Lors d'études du développement embryo-fœtal chez le rat, le sémaglutide a entraîné une embryotoxicité à des expositions inférieures aux niveaux cliniquement significatifs. Le sémaglutide a entraîné une nette réduction du poids maternel et une diminution de la croissance et de la survie embryonnaires. Chez les fœtus, des malformations viscérales et squelettiques majeures ont été observées, notamment des effets sur les os longs, les côtes, les vertèbres, la queue, les vaisseaux sanguins et les ventricules cérébraux. Des évaluations mécanistiques ont indiqué que l'embryotoxicité impliquait une anomalie médiée par les récepteurs du GLP-1 au niveau de l'apport de nutriments à l'embryon via le sac vitellin du rat. En raison des différences d'anatomie et de fonction du sac vitellin entre les espèces, et en raison de l'absence d'expression des récepteurs du GLP-1 dans le sac vitellin des primates non humains, ce mécanisme n'est probablement pas pertinent chez l'homme. Cependant, un effet direct du sémaglutide sur le fœtus ne peut être exclu.
Lors d'études de toxicité pour le développement chez le lapin et le singe cynomolgus, une augmentation des fausses couches et une légère hausse de l'incidence des anomalies fœtales ont été observées à des expositions cliniquement significatives. Ces résultats coïncidaient avec une nette réduction du poids maternel allant jusqu'à 16 %. Il n'est pas établi si ces effets sont liés à la réduction de consommation d'aliments par la mère en tant qu'effet direct du GLP-1.
La croissance et le développement postnataux ont été évalués chez le singe cynomolgus. Les nourrissons étaient légèrement plus petits à la mise-bas, mais ont récupéré pendant l'allaitement.
Chez les jeunes rats mâles et femelles, le sémaglutide a retardé la maturation sexuelle. Ces retards n'ont eu aucun impact sur la fertilité ou la capacité de reproduction des deux sexes, ni sur la capacité des femelles à maintenir une grossesse.
Wegovy
ne doit pas être utilisé si la solution n'est pas limpide et incolore.
Le stylo ne doit pas être utilisé s'il a été congelé.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Stylo prérempli, FlexTouch
Ce stylo est à usage multiple. Il contient 4 doses.
Il convient de recommander aux patients d'éliminer l'aiguille d'injection conformément à la réglementation en vigueur après chaque injection et de conserver le stylo Wegovy sans aiguille d'injection. Cela permet d'éviter une occlusion de l'aiguille, une contamination, une infection, une fuite de solution et des erreurs de posologie.
Le stylo ne doit être utilisé que pour une seule personne.
Wegovy peut être administré avec des aiguilles de 30G, 31G et 32G jetables de maximum 8 mm de long.
Liste I.
- Prescription initiale réservée aux spécialistes en
endocrinologie – diabétologie – nutrition ou aux médecins compétents en nutrition.
- Renouvellement non restreint.
Solution injectable (injection)
Solution isotonique, incolore et limpide ; pH = 7,4.
Stylo prérempli, FlexTouch (1,7 mg) en stylo prérempli de 3 ml
Cartouche en verre de 3 ml (verre de type I) fermée à une extrémité par un piston en caoutchouc (chlorobutyle) et à l'autre extrémité, par un bouchon en aluminium avec feuille de caoutchouc laminée (bromobutyle/polyisoprène) insérée. La cartouche est assemblée dans un stylo prérempli jetable en polypropylène, polyoxyméthylène, polycarbonate et acrylonitrile-butadiène-styrène.
Présentation : 1 stylo prérempli et 4 aiguilles NovoFine Plus jetables.
Chaque stylo prérempli contient 6,8
mg de sémaglutide* dans 3 ml de solution. Un ml de solution contient
2,27 mg de sémaglutide*. Un stylo prérempli contient 4 doses de 1,7 mg.
*peptide analogue au glucagon-1 humain (GLP-1) produit par la technique de l'ADN recombinant sur des cellules de Saccharomyces cerevisiae.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Phosphate disodique dihydraté
Propylène glycol
Phénol
Acide chlorhydrique (pour l'ajustement du pH)
Hydroxyde de sodium (pour l'ajustement du pH)
Eau pour préparations injectables
Commercialisation
de WEGOVY 1,7 mg, FlexTouch solution injectable en stylo prérempli,
boîte de 1 stylo prérempli (+ 4 aiguilles) de 3 ml (34009 3026024 1) le 08/10/2024
WEGOVY 1,7MG STYLO SC DOSES 4 (34009 5890349 6) reste disponible dans le cadre de l'accès précoce
Novo Nordisk
12 Cours Michelet
Carré Michelet
92800
Puteaux
Téléphone : 01 41 97 66 00
Information médicale
Tél
:
Ou 01 41
97 65 00 (Service et appel gratuits)
Fax : 
Email :
infomed@novonordisk.com
Site web : http://www.novonordisk.fr